【Cytiva】WB实验操作流程
Western blotting 标准流程
注意:使用去离子水或者双蒸水配制相关buffer。
1. 样本中加入loading buffer,加热样本至95°C,5分钟。
2. 上样前为去除聚集体或者颗粒,一般使用12 000 × g,2-5分钟对样品进行离心。
3. 将胶放在电泳仪中,加入适量的跑胶缓冲液。
4. 拔去凝胶中的梳子,让跑胶缓冲液没过每个上样孔。
5. 在样品孔中加入适量样品和彩虹marker。
6. 盖上电泳仪盖子,将电泳仪插孔插入电源,保证整个过程中黑对黑,红对红。
7. 跑胶。一般是浓缩胶80V,分离胶100V-120V。
8. 当溴酚蓝跑出凝胶下缘时,关掉电源,拆掉电泳装置取出凝胶板。
9. 去掉凝胶中的浓缩胶部分,将分离胶切角以记录凝胶方向。
接下来的步骤适用于湿转。
10. 在转印之前,将凝胶和PVDF膜放在预冷的转印缓冲液中平衡10至15分钟。
11. 将电转夹放入装有预冷的转移缓冲液托盘中,组装转移三明治,使蛋白质向膜迁移。
12. 将预先润湿的海绵放置在电转夹中,玻璃棒轻轻按压以除去气泡。
13. 将三张预先润湿的滤纸放在海绵上,玻璃棒轻轻按压以除去气泡。
14. 将膜放在滤纸上,玻璃棒轻轻按压以除去气泡。
15. 将凝胶放在膜的上面。在凝胶上放置另外三张预湿的滤纸。玻璃棒轻轻按压以除去气泡。
16. 最后将预先润湿的海绵放在最上面,玻璃棒轻轻按下以除去气泡,最后将夹子加紧。
17. 将预冷的转移缓冲液添加到转印装置中。
建议!在转印过程中添加磁搅拌子使缓冲液循环。
18. 将三明治的架子放入转移装置中。将转移装置连接到电源,转印通常是100V,1h。(转印具体参数需要根据蛋白特性和仪器说明书进行。)
19. 转移后,在适当的封闭溶液中将膜在室温下封闭1小时。
20. 封闭后,进行一抗孵育,通常在室温1h或4°C过夜。
21. 孵育后,需要对膜进行洗涤。使用含有Tween-20的磷酸盐缓冲盐水(PBS-Tween)或含Tween-20的Tris缓冲盐水(TBS-Tween)对膜进行洗涤,通常情况下是,每次洗涤5min,洗涤3次。
22. 洗涤完后,在室温下进行二抗体孵育1h。
23. 二抗孵育完后,通常在PBS-Tween或TBS-Tween中将膜洗涤3次,每次5分钟。
24. 根据所选检测系统的说明书进行蛋白质检测。
样品制备
㈠哺乳动物蛋白质提取
以下操作步骤我们以GE Mammalian Protein Extraction Buffer为例来介绍。这是一种特别适用于从哺乳动物细胞中提取蛋白质的溶液,适用于粘附细胞和悬浮细胞。
建议:
根据应用,可以将二硫苏糖醇(DTT)和乙二胺四乙酸(EDTA)加入缓冲液中。
在提取过程中添加蛋白酶抑制剂混合物。使用预冷的缓冲液并在冰上工作。
A、细胞悬液中蛋白的提取
1. 3000×g5分钟离心,去除并丢弃上清液。
2. 用5至10 mL预冷的PBS洗细胞一次。离心去除PBS清洗液。
3. 加入Mammalian Protein Extraction Buffer并悬浮细胞。对于每10mL悬浮物,加入1mLMammalian Protein Extraction Buffer。或者,每0.05g细胞加入1mLMammalian Protein Extraction Buffer。
4. 用移液管悬浮细胞,直到达到均一的悬浮液。将裂解物在冰上孵育15至30分钟。期间定期摇动或短暂涡旋悬浮液。
5. 在冷冻离心机中以20000×g离心裂解物30分钟。收集裂解液用于下游处理和分析。
B、铁壁细胞蛋白的提取
1. 对于贴壁细胞,首先弃掉培养基。
2. 用PBS洗涤细胞一次。弃掉PBS。
3. 加入适量的Mammalian Protein Extraction Buffer以覆盖细胞培养表面(表1)。或者,确保细胞可以从培养板上被刮下,
4. 并在后续步骤中将细胞作为悬浮细胞处理。
表 1 提取贴壁细胞需要用Mammalian Protein Extraction Buffer的体积
Buffer体积/每孔(μL) | 细胞培养板的类型 |
50 to 100 | 96孔板 |
100 to 200 | 24孔板 |
200 to 400 | 6孔板 |
250 to 500 | 60 mm 直径细胞培养皿 |
500 to 1000 | 100 mm 直径细胞培养皿 |
5. 轻轻摇动培养板10分钟,接着使用细胞刮将细胞轻轻地从平板上取下。裂解物,包括细胞碎片,可直接使用。或者,将裂解物转移至离心管中,以20000×g离心30分钟。收集澄清的裂解液用于下游处理和分析。
㈡样品除杂 SDS-PAGE Clean-Up Kit
SDS-PAGE除杂试剂盒用于制备由于高导电或低蛋白浓度而难以分析的SDS聚丙烯酰胺凝胶电泳(SDS-PAGE)样品。
该方法的工作原理是定量沉淀蛋白质,同时将干扰物质如洗涤剂、盐、脂类、酚类和核酸留在溶液中。
然后将蛋白质与SDS-PAGE loading buffer混合重新并重悬。该操作可在2小时内完成,定量收率高。
该试剂盒可以处理50个样品且每个样品的含量为100μl。该试剂盒可以扩大到更大体积或更稀释的样品。
实验所需要的材料:
· 1.5毫升离心管
· 冷冻离心机,可以提供 12000×g及以上的动力。
· 旋涡混合器
· β-巯基乙醇或DTT
· 95°c样品变性金属浴
前期准备:
开始操作前,将清洗缓冲液置于-20°C至少1 h。清洗缓冲液可储存在-20°C的冷冻室中。
向sample loading buffer中添加还原剂。还原剂可以是DTT或β-巯基乙醇。如果使用DTT,每100μl SDS-PAGE sample loading buffe添加3.1 mg,并确保完全溶解。如果使用β-巯基乙醇,每100μl sample loading buffer添加5μl。加入还原剂后,应立即使用sample loading buffer。或者可将sample loading buffer,可在-20°C下进行分装和储存。
蛋白样品应不含颗粒物质。必要时通过离心澄清。
在1.5 ml微量离心管中处理蛋白质样品。除非另有规定,否则所有步骤均应在冰上进行。将离心管放置在离心机中,使离心管盖子朝外。这样,样品将始终位于管的同一侧,最大限度地减少损失。
具体操作步骤:
1. 将1至100μl蛋白样品(含1μg至1 mg蛋白质)转移到1.5 ml微量离心管中。
2. 加入300μl沉淀剂(标为“1”),旋涡或反转混匀。在冰上孵育15分钟。
3. 在蛋白质和沉淀剂的混合物中加入300μl共沉淀剂(标记为“2”)。短暂的涡流混合。
4. 以最大速度(至少12000×g)在离心机中对管进行离心5分钟。离心完成后立即将管从离心机中取出。管壁上小颗粒应该是可见的。快速进行下一步,以避免颗粒再悬浮或分散。
5. 小心移液尽可能除去上清液。不要搅动沉淀。
6. 小心地将管子重新放置在离心机内,使离心盖子统一朝外。再次对管子进行离心,短暂的离心即可。用微量移液管除去剩余的上清液,使得管子中不应残留可见液体。
7. 用移液管将25μl水加到每个颗粒的顶部。每管旋转5到10秒。颗粒应分散,但不溶于水。
8. 添加1ml洗涤缓冲液(标记为“3”),在-20°C下放置至少1小时,以及5ul洗涤添加剂(标记为“4”)。旋涡直到颗粒完全分散。
9. 将离心管在-20°C下孵育至少30分钟。每10分钟旋涡20到30秒一次。
10. 以最大速度(至少12000×g)在离心机中离心管5分钟。
11. 小心地取出并丢弃上清液。一个白色的颗粒应该是可见的。让颗粒短暂风干(不超过5分钟)。干燥时间不要超过5分钟,如果太干,很难再悬浮。
12. 用5至40μl缓冲液I重悬颗粒(标记为“5”)。短暂涡流,在冰上孵育5分钟。
13. 步骤12中使用l缓冲液I每5μ添加1μl缓冲液II(标记为“6”)。短暂涡流,在冰上孵育5到10分钟。
14. 添加等量(6至48μl)的加入还原剂(DTT或β-巯基乙醇)(见前期准备)SDS-PAGE样品缓冲液(标记为“7”),如果溶液变黄,则以0.5μl的增量添加缓冲液I,直到溶液变蓝。
15. 将样品颠倒混匀5至10 s,在室温下孵育5至10 min。将样品管置于95°C金属预中煮3 min。
16. 对管进行短暂的离心,短暂即可。轻敲管子,确保内容物混合均匀。样品现在可以准备上样了。用GE公司的2-D Quant Kit试剂盒可以准确测定样品中的蛋白质浓度。
㈢使用2-D Quant试剂盒测定蛋白浓度
2-D Quant 试剂盒是专门为在电泳前准确测定样品中蛋白质浓度而设计的。该分析对体积在1到50μl之间的高达50μg的可溶性蛋白质。
具有线性响应。该方法与常用的样品制备试剂(如2%SDS、1%DTT、8 M尿素、2 M硫脲和4%的3-[(3-胆酰胺丙基)二甲基氨]-1-丙磺酸酯(Chaps)兼容,给你的蛋白定量准确性保驾护航。
实验所需要的材料:
--沉淀剂、共沉淀剂、铜溶液、彩色试剂A和B,和牛血清白蛋白(BSA)标准溶液(试剂盒提供的。)
--2ml离心管
--旋涡混合器
--离心机
--可见光光度计
具体操作步骤:
1. 制备适量的工作颜色试剂,将100份色剂A与1份色剂B混合。
每个单独的分析需要1毫升工作颜色试剂。工作颜色试剂可在4°C至8°C下储存1 周或只要溶液的光密度(OD)在480 nm下保持在0.025以下都可以。
2. 将BSA标准溶液(2 mg/ml)加入6个试管中,制备标准曲线,最终量为:0(空白)、10、20、30、40和50μg。
3. 制备待分析样品。在单独的试管中添加1至50μl待分析样品。调整此体积,确保在分析的工作范围内添加一定量的蛋白质(0.5至50μg)。
4. 每管(包括标准曲线管)加沉淀剂500μl。短暂旋转,在室温下静置2到3分钟。
5. 在每管中加入500μl共沉淀剂,通过旋涡或反转进行短暂混合。
6. 对试管进行至少10000×g离心5分钟。使用tip头丢弃上清液。建议增加一个短暂离心,然后用微量tip头除去任何残留的上清液。
7. 每管加入100μl铜溶液和400μl水。短暂旋涡以溶解沉淀的蛋白质。
8. 每管加100μl工作颜色试剂。尽可能快地加入试剂,确保即时混合。颠倒混匀,室温孵育15-20分钟。
9. 以水为参考,在480 nm处读取每个样品和标准品的吸光度。吸光度应在加入工作色试剂后40分钟内读取完毕。
10. 通过绘制标准品对蛋白质量的吸光度,生成标准曲线。用这个标准曲线来测定样品的蛋白质浓度。
电泳
通用PAGE(聚丙烯酰胺凝胶电泳)实验步骤:
1. 加入sample loading buffer,在95°C下煮样5分钟。
对于变性和还原条件下的PAGE:向sample loading buffer中添加SDS和还原剂。
对于变性条件下的PAGE:向sample loading buffer添加SDS。
对于 Native胶:无SDS,无还原剂,无加热!
样品加入sample loading buffer煮沸后,可以在-20°C下保存3至4周或在4°C下保存1 周。在分析前将样品加热至37°C几分钟,以使沉淀的SDS再溶解。
上样前,务必检查蛋白质浓度。
2. 将凝胶放入电泳设备中,加入适当的缓冲液。
3. 取下梳子,使缓冲液没过上样孔。
4. 将样品和marker加入到凝胶孔中。
上样前,将所有样品离心12000×g 2至5分钟,以去除任何颗粒物。
建议用sample loading buffer补满未加入样品或者marker的泳道。
5. 将盖子盖上,并将插头黑对黑红对红插入电源插座中。
6. 在适当的条件下跑电泳。一般情况下, 80V浓缩胶,120V分离胶。
7. 当染料到达凝胶底部时,关闭电源,断开导线,取下盖子。
8. 将凝胶从电泳装置中取出,进入下一步。
㈠SDS-PAGE胶的制备
制胶过程中用的 buffer:
--4× resolving gel buffer (1.5 M Tris-Cl, pH 8.8):
--4× stacking gel buffer (0.5 M Tris-HCl, pH 6.8)
--10% SDS
--10% ammonium persulfate (APS)
制胶的基本流程:
1. 在制胶单元上组装玻璃板,使用适当厚度的垫片来确定最后的上样量。
2. 在玻璃瓶中依次加入制作分离胶所需的各项缓冲液(见表2),先不要加入APS和TEMED。
3. 使用tip头反复吹吸,使得各项缓冲液混匀,尽可能不要引入气泡。
4. 加入Temed和APS,轻轻旋转玻璃瓶,注意不要产生气泡。
5. 用移液管将混好的分离胶加入到胶板中。
6. 在分离胶上立即加入纯水或异丙醇或乙醇,将凝胶压平。当凝胶聚合时,可以看到非常尖锐的液体-凝胶界面。
7. 在玻璃瓶中依次加入各项buffer(见表3)制备浓缩胶,先不加APS和TEMED。
8. 待下层分离胶凝固后,将覆盖层纯水或异丙醇或乙醇倒掉。必要时可以用滤纸吸取残余液体。
9. 在玻璃瓶中加入50μl的10%APS和10μl的TEMED。轻轻混匀,注意不要产生气泡。
10. 用移液器将浓缩胶加在分离胶上方,注意不要产生气泡。
11. 小心地将梳子放入未聚合的凝胶中,静置等待凝胶凝固。
凝胶可与梳子一起存放,在4°C的密封袋内用塑料包装紧紧包裹,保持1 周。保持凝胶湿润。
表2 分离胶,40ML
Final gel concentration | 4% |
丙烯酰胺溶液 | 1.33 mL |
4× 浓缩胶缓冲液 | 2.5 mL |
10% SDS | 0.1 mL |
水 | 6 mL |
TEMED1 | 5 μL |
表3 浓缩胶溶液, 10 mL
最终胶浓度 | 5% | 7.5& | 10% | 12.5% | 15% |
丙烯酰胺溶液 | 6.7 mL | 10 mL | 13.3 mL | 16.7 mL | 20 mL |
4× 分离胶缓冲液 | 10 mL | 10mL | 10mL | 10mL | 10mL |
10% SDS | 0.4mL | 0.4 mL | 0.4 mL | 0.4 mL | 0.4 mL |
水 | 22.7mL | 19.4mL | 16.1 mL | 12.8 mL | 9.5 mL |
10% APS1 | 200 μL | 200 μL | 200 μL | 200 μL | 200 μL |
TEMED1 | 13.3 μL | 13.3 μL | 13.3 μL | 13.3 μL | 13.3 μL |
转印
㈠湿转
所需材料:
· 电泳后聚丙烯酰胺凝胶
· PVDF膜或者NC膜
· 四片WHWMAN×3毫米滤纸,切割成与凝胶相同的尺寸
· 两个泡沫垫
· 转印模块,电源
· 100%甲醇
· 水
· 转移缓冲液:25 mM Tris base, 192 mM glycine, ≤ 20% (V/V) methanol, pH 8.3 在使用前,转移缓冲液需预冷。
转印步骤:湿转
1. 准备足够的转移缓冲液,转印缓冲液需提前预冷。
2. 电泳后,凝胶切角以辨认方向。
3. 将凝胶在转移缓冲液中平衡10至15分钟。记住,凝胶的不完全平衡可能导致条带弥散。
4. 根据制造商的建议,对膜进行裁剪、预湿和平衡。
对于PVDF膜,需在甲醇中预湿15 s,然后用水润湿。在转印缓冲器中平衡至少10分钟。
对于NC膜,在转移缓冲液中平衡至少10分钟。
5. 在转移缓冲液中浸泡滤纸和泡沫垫。
6. 将转印盒放在托盘中,托盘中预先加入转印缓冲液。
7. 转印盒从阳极(+)侧开始,依次放入泡沫海绵,两个滤纸,制备的膜,凝胶,两个湿滤纸,第二个泡沫海绵。
确保滤纸、凝胶和膜之间有气泡。
8. 在转印装置中放置搅拌子。
9. 将夹紧的转印三明治,确保无气泡,放在转印装置中,黑对黑,红对红。
10. 将转印装置黑对黑红对红接到电源上。
11. 根据制造商的建议进行转移。通常是恒压100V 1h。
12. 转移后,从转印三明治中取下薄膜,从薄膜上切一个角以便定位,然后在PBS或TBS中短暂冲洗。
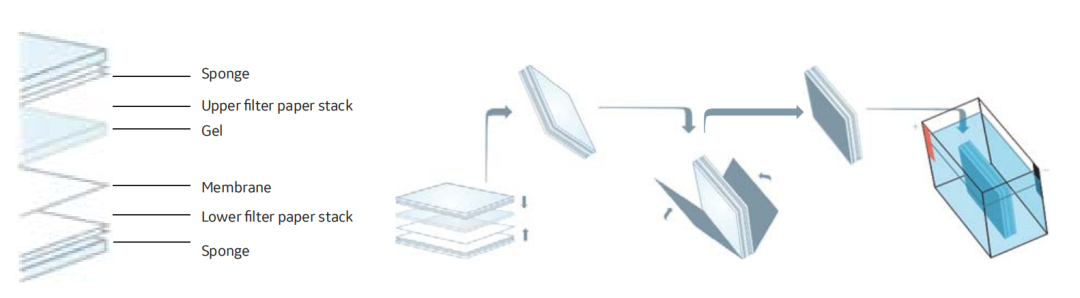
图1 湿转系统
㈡半干转
所需材料:
· 电泳后的聚丙烯酰胺凝胶电泳
· PVDF膜或者NC膜
· 至少6个3毫米滤纸,切成与凝胶相同的尺寸
· 半干转印仪、电源
· 100%甲醇
· 水
· 转移缓冲液:25 mM Tris base, 192 mM glycine, ≤ 20% (V/V) methanol, pH 8.3 在使用前,转移缓冲液需预冷。
转印步骤:半干转
1. 用水冲洗半干转印仪的阳极(+)和阴极(-)。
2. 电泳后,凝胶切角以辨认方向。
3. 从凝胶中除去浓缩胶和溴酚蓝染料前端。
4. 将凝胶在转移缓冲液中平衡10至15分钟。记住,不完全平衡的溶解凝胶可能导致条带弥散。
5. 将至少六片滤纸切成与凝胶相同的尺寸或稍小。
6. 用转移缓冲液浸泡至少三张滤纸。滤纸一个接一个置于下电极(阳极[+])的中心,然后用干净的吸液管或滚筒从
中心向边缘滚动,以除去所有的空气。
7. 将膜切割成与凝胶相同的尺寸或稍小。预湿并在转移缓冲液中平衡膜至少10分钟。
在转移缓冲液中平衡之前,PVDF膜必须在甲醇中预湿并在水中冲洗。在平衡之前,硝化纤维素膜应在水中预先湿润。处理膜时务必戴上干净的手套,以避免留下指纹。
8. 将预湿膜放在滤纸上。
9. 将凝胶放置在膜上。然后用干净的吸液管或滚筒从中心向边缘滚动,以除去所有的空气。
10. 用三层湿滤纸覆盖凝胶。小心堆叠每层,边缘平行。每层添加后,从中心到边缘滚动干净的吸液管,去除所有的气泡。
11. 将转印装置上层盖上,将彩色导线连接到电源上。黑对黑,红对红。
12. 根据制造商的建议设置半干传输通常在恒定电流下进行。转印时间通常约为1小时。
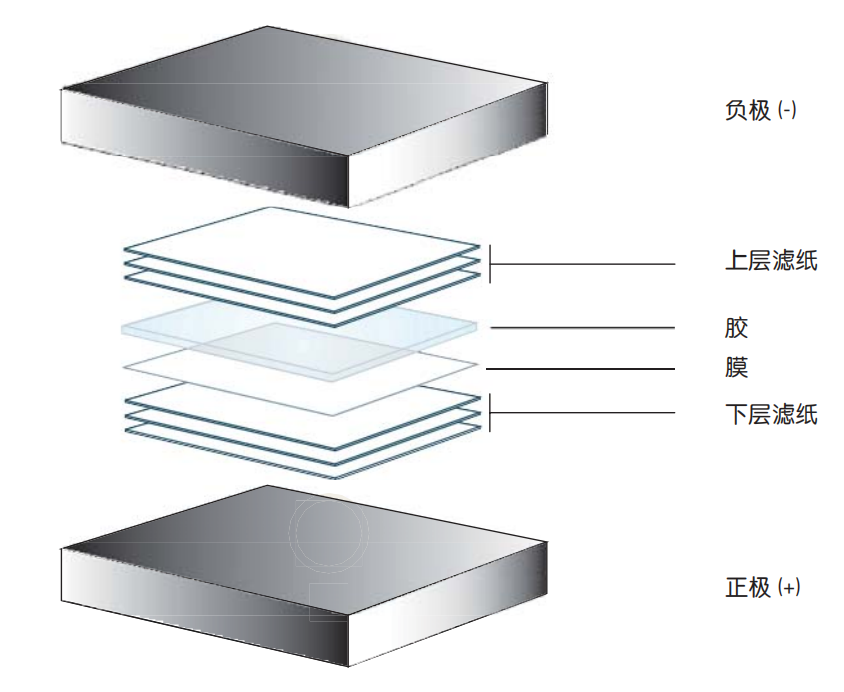
图2 半干转系统
封闭
如果蛋白检测目标本身是磷酸化,不要使用PBS作为blocking稀释缓冲液-使用TBS。
如果使用生物素化或刀豆球蛋白结合作为抗体检测,不要使用脱脂牛奶作为封闭剂。
如果蛋白检测目标物被磷酸化,不要使用粗蛋白制剂作为封闭剂。
注意,并非所有的封闭剂都与荧光蛋白印迹法兼容。Amersham ECL Prime Blocking Agent 建议可以用于荧光检测western。
具体操作步骤:
1. 摇动粉末块,确保成分均匀。
2. 称出适当量的封闭剂,配置溶液使得浓度为2%(w/v) (Amersham ECL Prime Blocking Reagent) 或5% (w/v) (Amersham
ECL Blocking Reagent)。
3. 加入适量PBST或TBST,用力摇动,搅拌15分钟,直到所有成分完全溶解。
制备的封闭溶液可在2°C至8°C下储存,但应在24小时内使用。
3. 将膜置于封闭溶液中,在室温下孵育1h,如果背景持续过高,可以尝试在37°C下孵育1h。或者,如果方便的话,膜可以在2°C到8°C的温度下在封闭溶液中过夜。
5. 在清洗缓冲液中短暂冲洗膜。
抗体孵育和检测
㈠化学发光(以Amersham ECL, Amersham ECL Prime, and Amersham ECL Select为例)
1. 用PBST/TBST稀释一抗,具体稀释倍数需要经过优化。
2. 将膜(蛋白质面朝上)放入一抗溶液中,并在室温下孵育1 h或者4℃过夜,请参考抗体说明书。
3. 在PBST/TBST中清洗膜三到六次,每次清洗5分钟。
4. 将膜置于PBST/TBST稀释的二抗中,通常在室温下孵育1h,请参考抗体说明书。
5. 将膜放入PBST/TBST中,每次洗涤4至6次,每次洗涤5分钟。
6. 按照所选检测试剂和成像系统的建议继续检测。
基于Ai680 CCD成像系统
1. 让检测溶液平衡到室温。
2. 混合等量的检测溶液A和B,使总体积足够覆盖膜。一般需要0.1 ml/cm2的膜体积。
3. 将膜从洗涤液中拿出,并将其蛋白质面朝上放在检测托盘内。用移液管将混合检测试剂加到膜上。
4. 在室温下孵育1分钟(Amersham ECL)或5分钟(Amersham ECL Prime和Amersham ECL Select)。
5. 将样品盘放入Ai680成像仪中,按说明书操作。选择曝光时间并拍摄图像。
根据预期信号强度选择曝光时间。建议从1分钟开始,然后调整时间以找到最佳曝光。或者,可以使用累积曝光,可以固定时间叠加一定数量的图片。
㈡荧光检测(Amersham ECL Plex)
1. 在洗涤液或封闭液中稀释小鼠或兔源性一抗至最佳浓度。
2. 用稀释的一抗孵育膜(蛋白面朝上)室温下1.5小时,或4°C下过夜。
3. 在室温下洗膜,每次5分钟,共洗3次。
4. 稀释ECL-Plex-CyDye结合二抗(以1μg/ml的浓度制备)至最佳浓度。
5. 将清洗过的膜在二抗溶液中在室温下孵育1h。确保薄膜不受光照。
6. 在室温下洗膜,每次5分钟,共洗3次。保护膜不受光照。当使用荧光标记抗体时,应在避光处理。
7. 用激光扫描仪扫描膜,检测二抗信号。
㈢多通道荧光检测
1. 一抗使用PBST或TBST稀释。
在单一溶液中可混合一种以上抗体以进行多重检测。
为了防止串扰,不同种一抗必须来源于不同物种。如果使用化学发光检测,则对感兴趣的蛋白质必须在电泳中进行
很好的分离。
如果一抗要重复使用,建议一抗还是分别孵育。
2. 将膜(蛋白质面朝上)放入一抗溶液中,室温下孵育1h或4°C下过夜。
3. 在室温下洗膜,每次5分钟,共洗3次。
4. 稀释二抗,具体比例需要优化。
5. 将膜置于混合的二抗溶液中,在室温下培养1h,保护膜不受光照。
当使用荧光标记抗体时,应在黑暗中孵育。
6. 在室温下洗膜,每次5分钟,共洗3次。保护膜不受光照。
7. 用荧光激光扫描仪扫描细胞膜,检测二抗信号。
剥离和再杂交
如果计划剥离和再杂交,PVDF膜比NC膜更坚固,因此建议使用NC膜。
㈠在高PH和高温下进行剥离
1. 将膜浸入剥离缓冲液(100 mM β-巯基乙醇,2%SDS,62.5 mM Tris-HCl【ph 6.7】),在70°C下孵育30分钟,期间不停搅动。
较低的温度(50°C到70°C)也可能很好地工作,但这应该通过使用抗体的经验来确定。
2. 用PBST或TBST在室温下用大量洗涤液洗涤膜两次,每次洗涤10分钟。
3. 在室温下,在合适的封闭溶液中封闭膜1小时。
4. 重复孵育和检测。
㈡在低PH下进行剥离
1. 将膜浸入剥离缓冲液(100 mMβ-巯基乙醇,1%SDS,25 mM甘氨酸HCl[ph 2.0])中,孵育30分钟,期间不停搅动。
2. 用PBST或TBST在室温下用大容量洗涤液洗涤膜两次,每次洗涤10分钟。
3. 在室温下,在合适的封闭溶液中封闭膜1小时。
4. 重复孵育和检测。
㈢在高PH下进行剥离
1. 将膜浸入0.2 m NaOH中,在室温下孵育5分钟,期间不停晃动。
2. 取出并加入0.2 m的新鲜NaOH,再培养5分钟。
3. 用水冲洗5分钟。若仍有信号残留,则将NaOH浓度增加至2 M,培养时间增加至30 min。
4. 重复孵育和检测(一般无需封闭)。
使用NaOH剥离后通常不需要重新封闭。但是,根据NaOH浓度和浸泡时间,可能需要重新封闭。
㈣在高盐下进行剥离
1. 将膜浸泡在添加0.5 m NaCl和0.2%SDS的PBS或TBS中30分钟至2小时。
2. 用水冲洗膜。
3. 重复孵育和检测(一般无需封闭)。
使用盐剥离后通常不需要重新封闭。但是,根据浸泡时间的不同,可能需要重新封闭。
其他细胞品类

别划走,精彩继续

长按识别
添加一对一专属技术支持

扫码进群
咨询详细产品信息

点在看,传递你的品味