分子伴侣介导的自噬导致 PLCG1 降解受损, 从而促进细胞衰老和椎间盘退变

分子伴侣介导的自噬(CMA)缺陷与细胞衰老相关,但具体机制尚不明确。本研究中发现CMA抑制会以钙依赖的方式诱导细胞衰老,并明确了其在肿瘤坏死因子(TNF)诱导的髓核细胞(NPC)衰老及椎间盘退变中的作用。
1. 椎间盘退变(IVDD)是慢性下腰痛的主要诱因,核心病理为炎症因子(如TNF)诱导髓核细胞(NPC)衰老。
2. CMA(分子伴侣介导的自噬)缺陷与细胞衰老相关,但二者在IVDD中的关联及机制不明;钙超载是NPC衰老和IVDD的重要触发因素,其上游调控机制尚未明确。
3. CMA失调、钙超载、细胞衰老是IVDD及多种年龄相关退行性疾病的共同特征,但连接三者的关键分子靶点尚未发现。
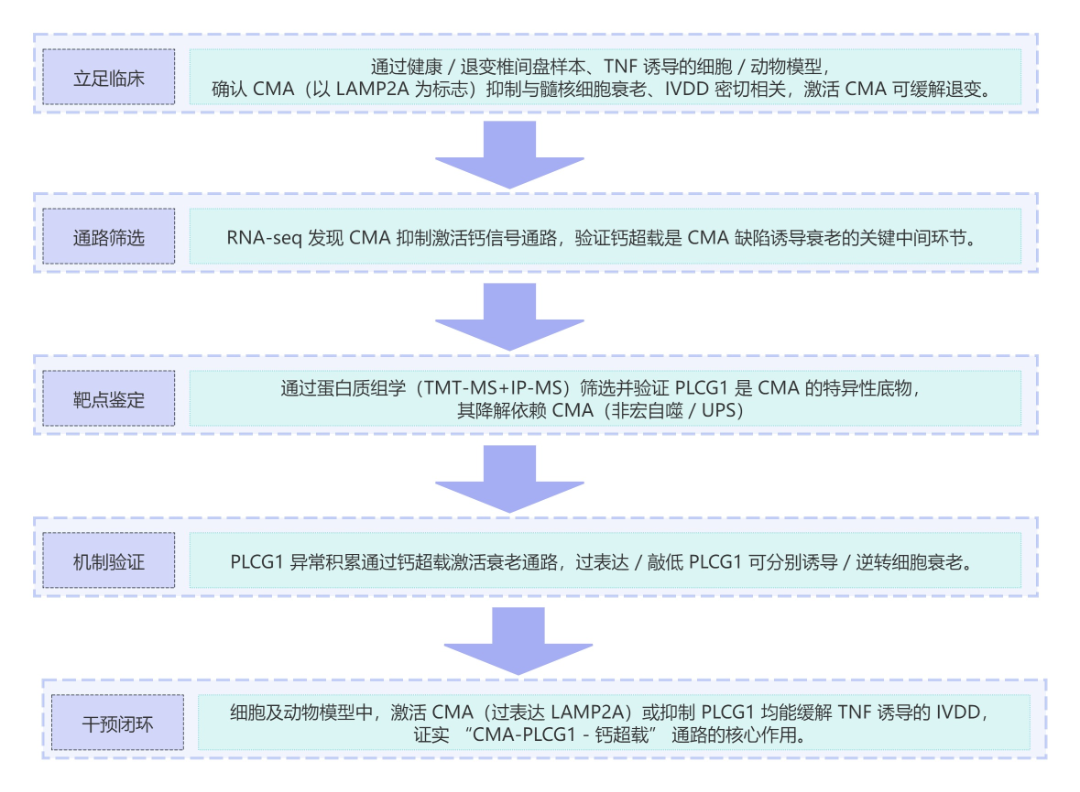
以CMA缺陷介导椎间盘退变(IVDD)的机制为核心,先通过临床样本、细胞及动物模型,证实CMA抑制与髓核细胞衰老、IVDD密切相关。
再经蛋白质组学筛选出CMA特异性底物PLCG1,验证其积累会引发钙超载,进而诱导细胞衰老。
最后通过体内外实验确认,激活CMA(过表达LAMP2A)或抑制PLCG1,均可缓解TNF诱导的IVDD,阐明“CMA-PLCG1-钙超载”的核心调控链。
1. 分子伴侣介导的自噬(CMA)减轻肿瘤坏死因子(TNF)诱导的细胞衰老及椎间盘退变(IVDD)。
CMA在脑、肝等多器官及椎间盘(IVD)中具有活性,其关键分子LAMP2A在椎间盘的表达提示其可能参与椎间盘退变(IVDD)调控。
临床样本显示,退变椎间盘的LAMP2A(蛋白+mRNA)显著降低,衰老标志物升高。细胞实验证实,炎症因子TNF会抑制髓核细胞的CMA活性(LAMP2A下调、CMA特异性斑点减少),同时诱导细胞衰老;而CMA激活剂或血清剥夺可增强CMA活性。
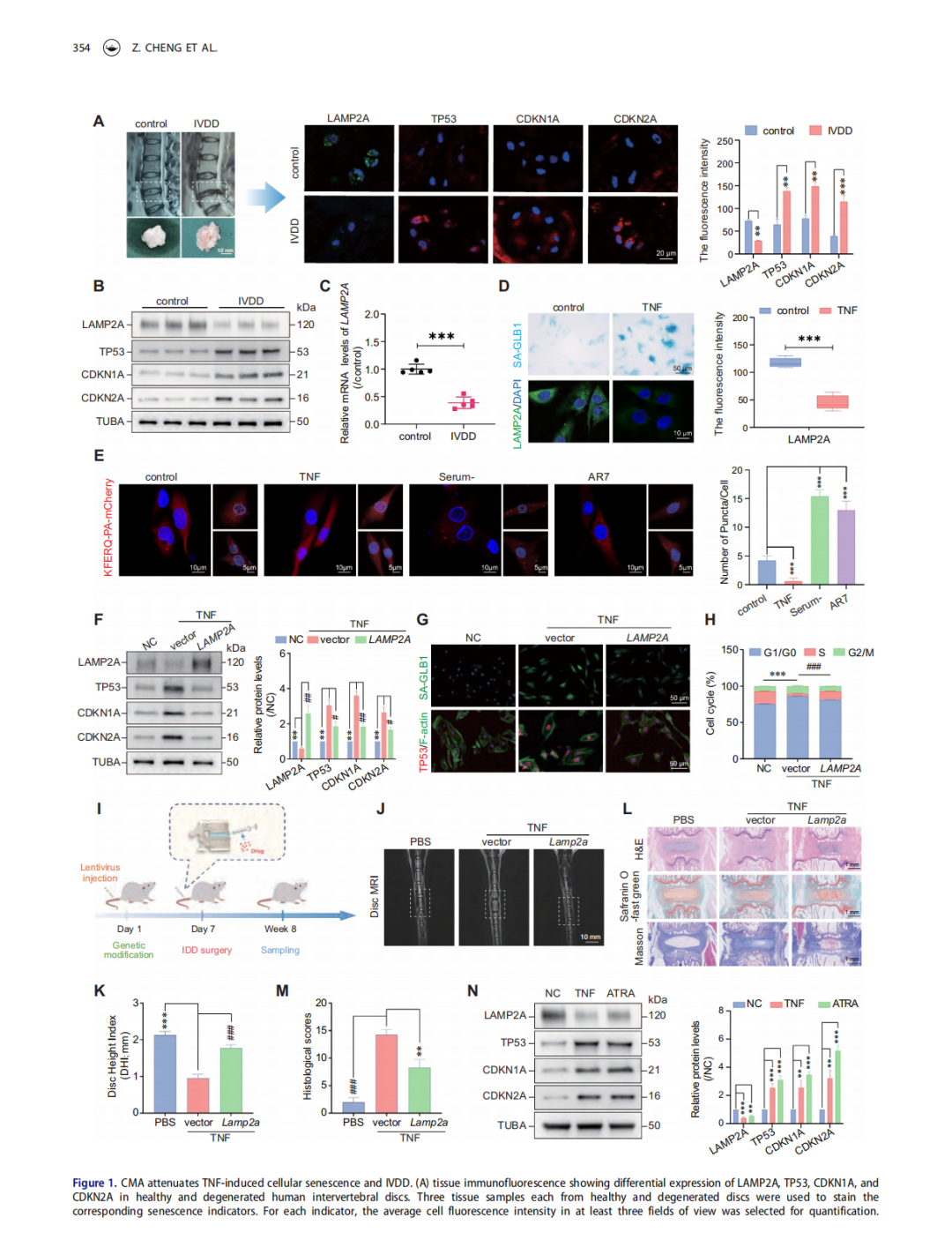
为验证TNF诱导的髓核细胞(NPC)衰老及椎间盘退变(IVDD)是否与CMA抑制相关,研究开展以下实验并得出结论:
细胞实验:慢病毒过表达LAMP2A后,TNF诱导的衰老标志物(TP53、CDKN1A等)上调、SA-GLB1激活及细胞周期阻滞(S期比例、BrdU水平降低)均被逆转。
动物实验:大鼠椎间盘内注射TNF联合LAMP2A过表达,8周后椎间盘退变程度显著减轻(尾僵硬缓解、Pfirrmann评分及组织学评分降低、椎间盘高度塌陷减少),且退变相关标志物上调受抑。
补充验证:CMA抑制剂ATRA处理NPC,同样诱导衰老标志物升高,与TNF作用一致。
综上,LAMP2A过表达(激活CMA)可在体内外减轻TNF诱导的NPC衰老及IVDD,证实CMA抑制介导了该病理过程。
2.CMA(分子伴侣介导的自噬)抑制促进钙超载诱导的细胞衰老
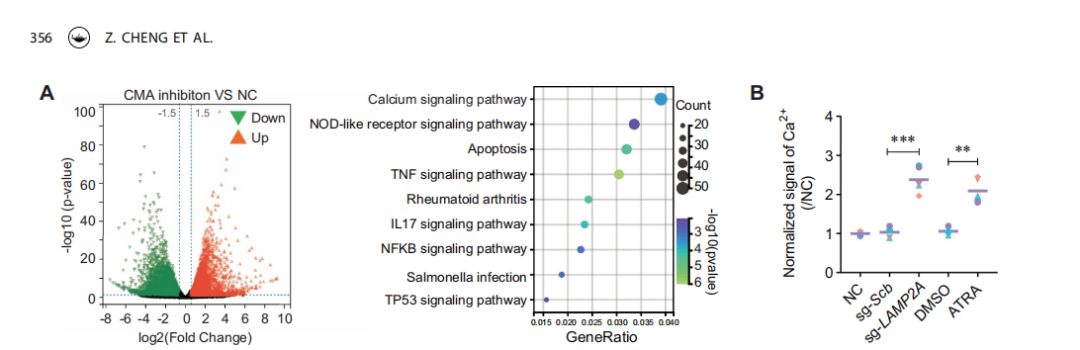
为探究CMA抑制影响细胞衰老的潜在机制,研究对CMA抑制剂处理后的髓核细胞(NPC)进行RNA测序。KEGG通路富集分析显示,CMA抑制显著激活钙信号通路,这一结果经后续Ca²⁺水平检测得到证实。
结合已知研究(内质网到线粒体的Ca²⁺流是细胞衰老的动力,钙超载是NPC衰老及椎间盘退变的触发因素),研究推测CMA抑制诱导的细胞衰老可能与钙超载相关,而CMA在钙稳态中的作用此前尚未被正式报道。
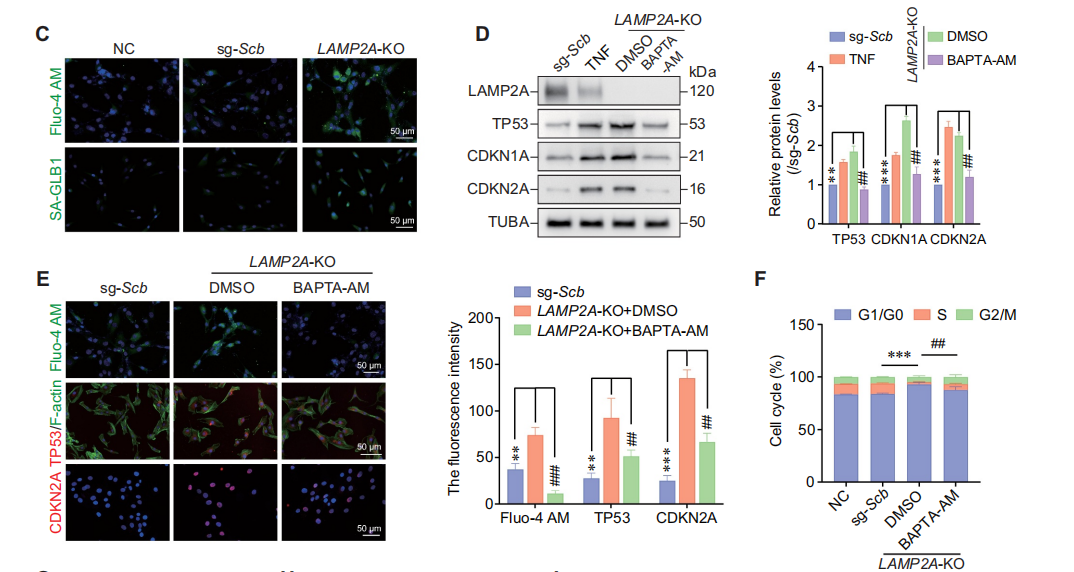
研究利用CRISPR-Cas9系统敲除人髓核细胞的LAMP2A基因,设计靶向LAMP2外显子9差异区的sgRNA并筛选出有效靶点sgRNA#3,该靶点可特异性敲除LAMP2A,且不影响LAMP2其他亚型及主要溶酶体蛋白酶的正常表达。
实验发现,LAMP2A敲除的髓核细胞中Ca²⁺水平显著升高,衰老标志物SA-GLB1上调,还出现S期比例降低、BrdU水平下降的细胞周期阻滞现象。而用钙螯合剂BAPTA-AM处理后,上述敲除引发的钙超载、衰老相关蛋白上调及细胞周期阻滞均被显著缓解。
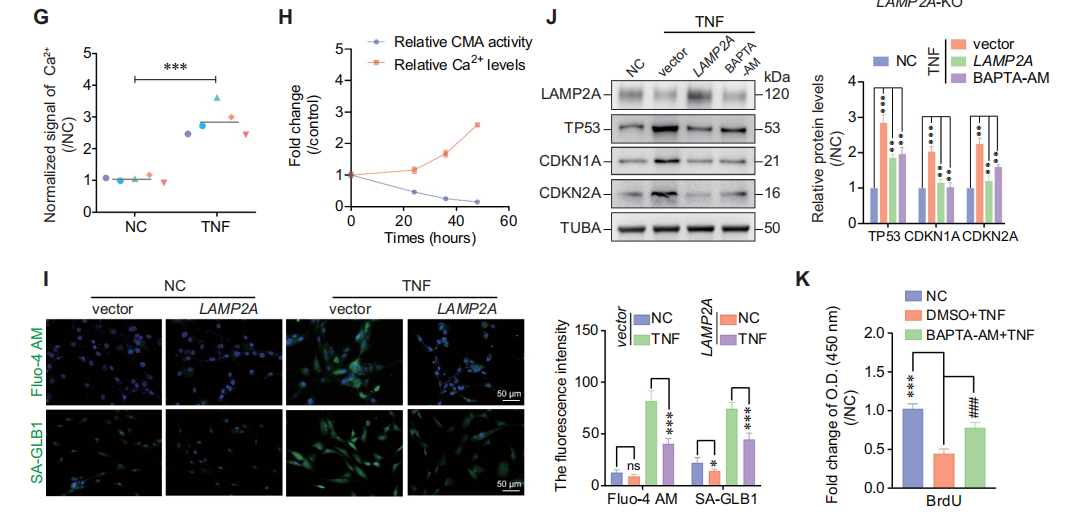
为明确CMA抑制引发的钙超载在TNF诱导髓核细胞(NPC)衰老中的作用,研究开展系列实验:
首先,检测发现TNF处理可升高NPC的Ca²⁺水平;追踪CMA活性与Ca²⁺水平变化显示,TNF处理24h时CMA已受抑但Ca²⁺无明显升高,36、48h时CMA抑制伴随Ca²⁺升高,表明CMA抑制先于钙超载,提示其介导TNF诱导的钙超载。
此外,慢病毒过表达LAMP2A(激活CMA)可使NPC抵抗TNF诱导的钙超载与衰老;过表达LAMP2A或用钙螯合剂BAPTA-AM处理,均能抑制TNF诱导的衰老标志物上调,且BAPTA-AM可逆转TNF诱导的细胞周期阻滞。
3.磷脂酶 Cγ1 被筛选为 CMA 的潜在底物
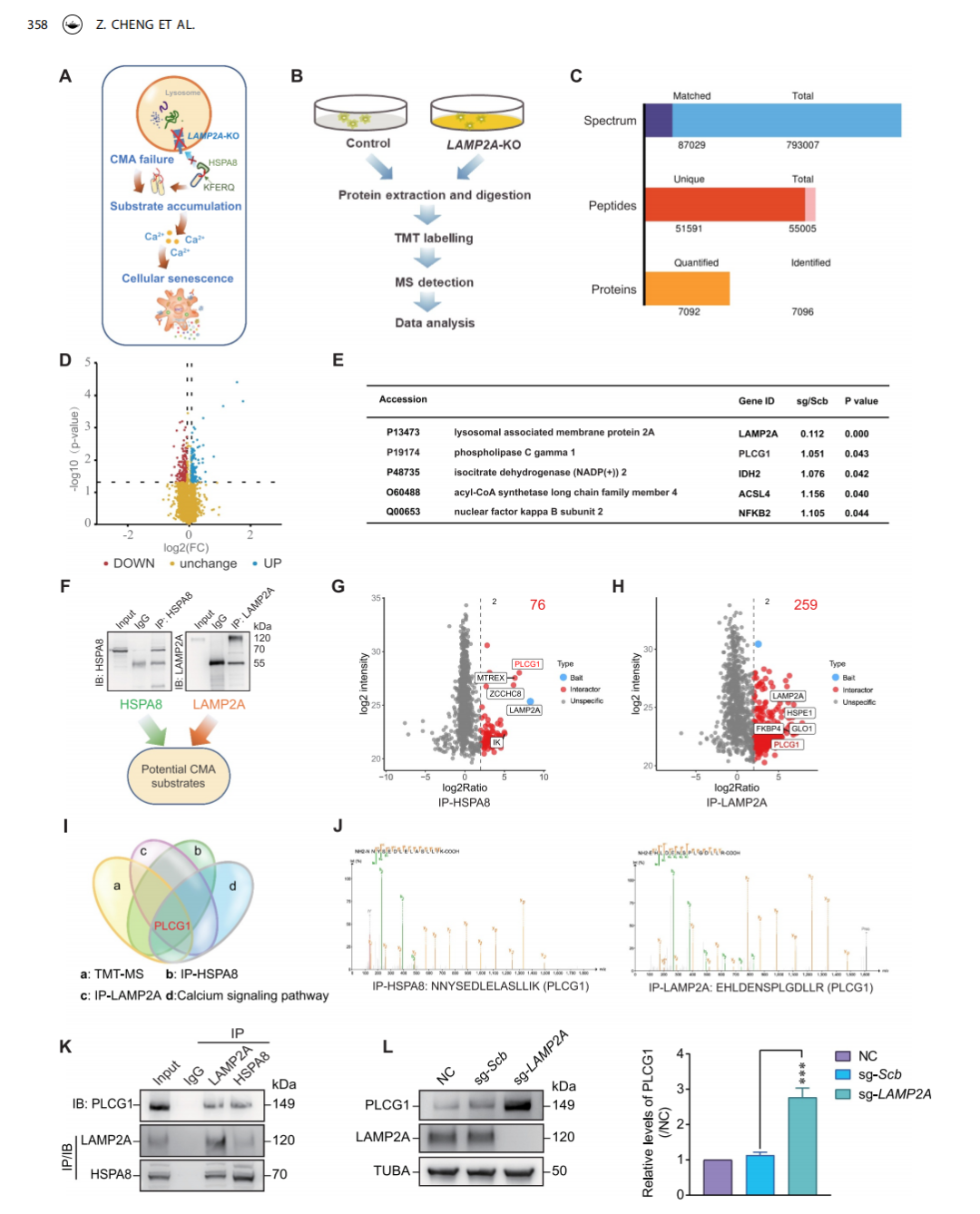
研究推测CMA抑制会因特定底物降解异常引发钙超载,遂依据CMA底物的特征(含KFERQ样序列、LAMP2A敲低后上调、与HSPA8/LAMP2A互作)筛选其调控钙稳态的潜在底物。
首先,构建LAMP2A敲除的髓核细胞并进行TMT标记定量质谱分析,验证了结果可靠性;再对HSPA8、LAMP2A做免疫沉淀结合无标记质谱,筛选二者高评分互作蛋白。
随后,整合TMT-MS结果、HSPA8与LAMP2A的共互作蛋白及钙信号通路基因集,经程序化基序特征筛选,锁定PLCG1为候选底物。
最后,验证了PLCG1与HSPA8/LAMP2A存在相互作用,且其在LAMP2A敲除的髓核细胞中表达显著升高,提示PLCG1可能是CMA抑制引发钙流异常的潜在介导因子。
4.磷脂酶 Cγ1(PLCG1)受分子伴侣介导的自噬(CMA)调控,而不受大自噬(宏自噬)或泛素 - 蛋白酶体系统(UPS)的调控。
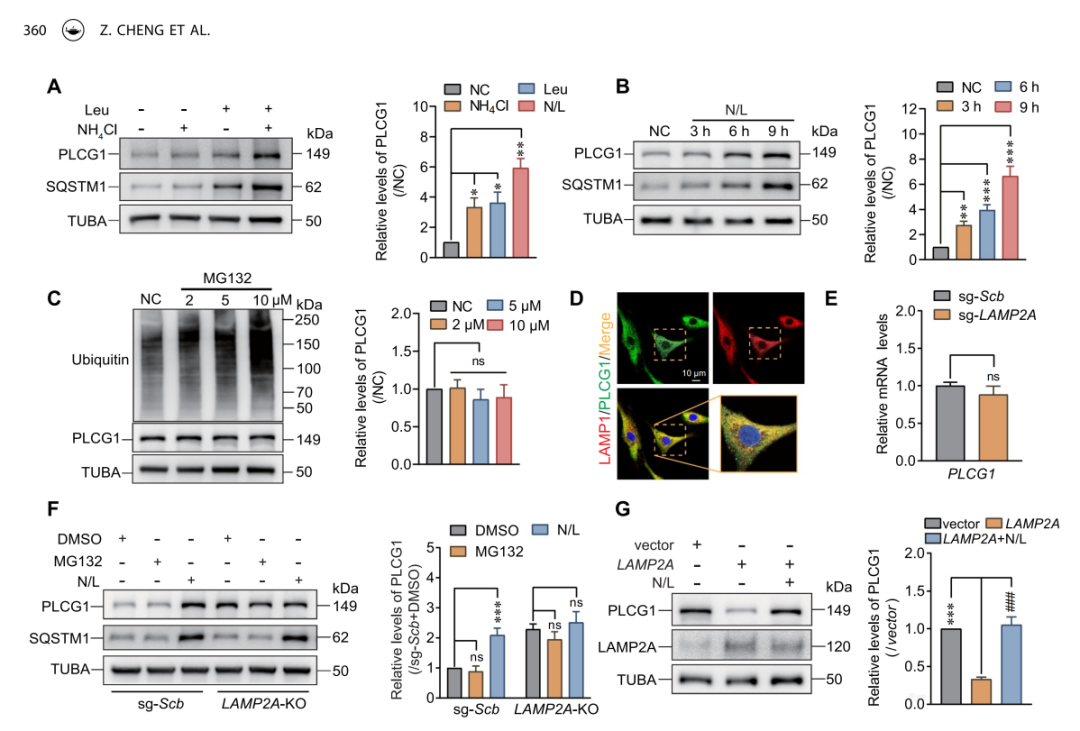
研究为探究PLCG1的降解途径开展系列实验:
首先用溶酶体抑制剂亮肽素(Leu)、氯化铵(NH₄Cl)及二者联用处理髓核细胞,发现其可抑制PLCG1降解,且抑制效果呈时间依赖性、可叠加,而泛素-蛋白酶体系统(UPS)抑制剂MG132对PLCG1表达无影响,免疫荧光也证实PLCG1与溶酶体共定位,说明PLCG1经溶酶体途径降解,而非UPS。
进一步实验显示,LAMP2A敲除后PLCG1蛋白水平升高但mRNA水平无变化,且敲除组中溶酶体、UPS抑制剂均无法影响PLCG1水平,提示PLCG1的溶酶体降解依赖LAMP2A。
最后,过表达LAMP2A激活CMA可降低PLCG1水平,而联用溶酶体抑制剂能逆转该效应,进一步验证PLCG1的降解受CMA(LAMP2A)调控。
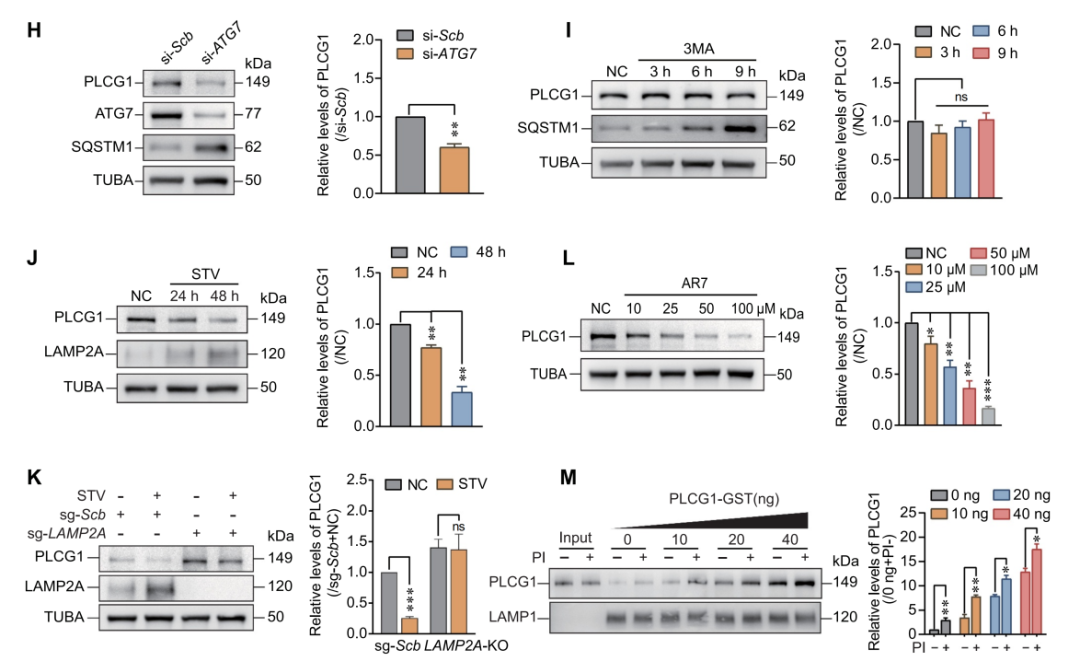
研究进一步验证PLCG1的降解仅受CMA调控,与宏自噬及UPS无关,具体实验及结果如下:
探究宏自噬的影响:敲低宏自噬关键分子ATG7抑制宏自噬(SQSTM1升高),但PLCG1水平未升反降;用宏自噬抑制剂3MA或激活剂雷帕霉素处理髓核细胞,均不影响PLCG1水平,说明宏自噬不调控PLCG1。
验证CMA的调控作用:血清剥夺(CMA激活条件)可使髓核细胞LAMP2A升高、PLCG1降低,且PLCG1与溶酶体共定位增强,而LAMP2A敲除会消除该效应;CMA激活剂(AR7、QX77等)可浓度依赖性降低PLCG1水平。
体外验证:GST-PLCG1与纯化的活性溶酶体共孵育会被降解,且证实HSPA8与PLCG1存在内源性互作。
5.磷脂酶 Cγ1(PLCG1)是分子伴侣介导的自噬(CMA)的确证底物。
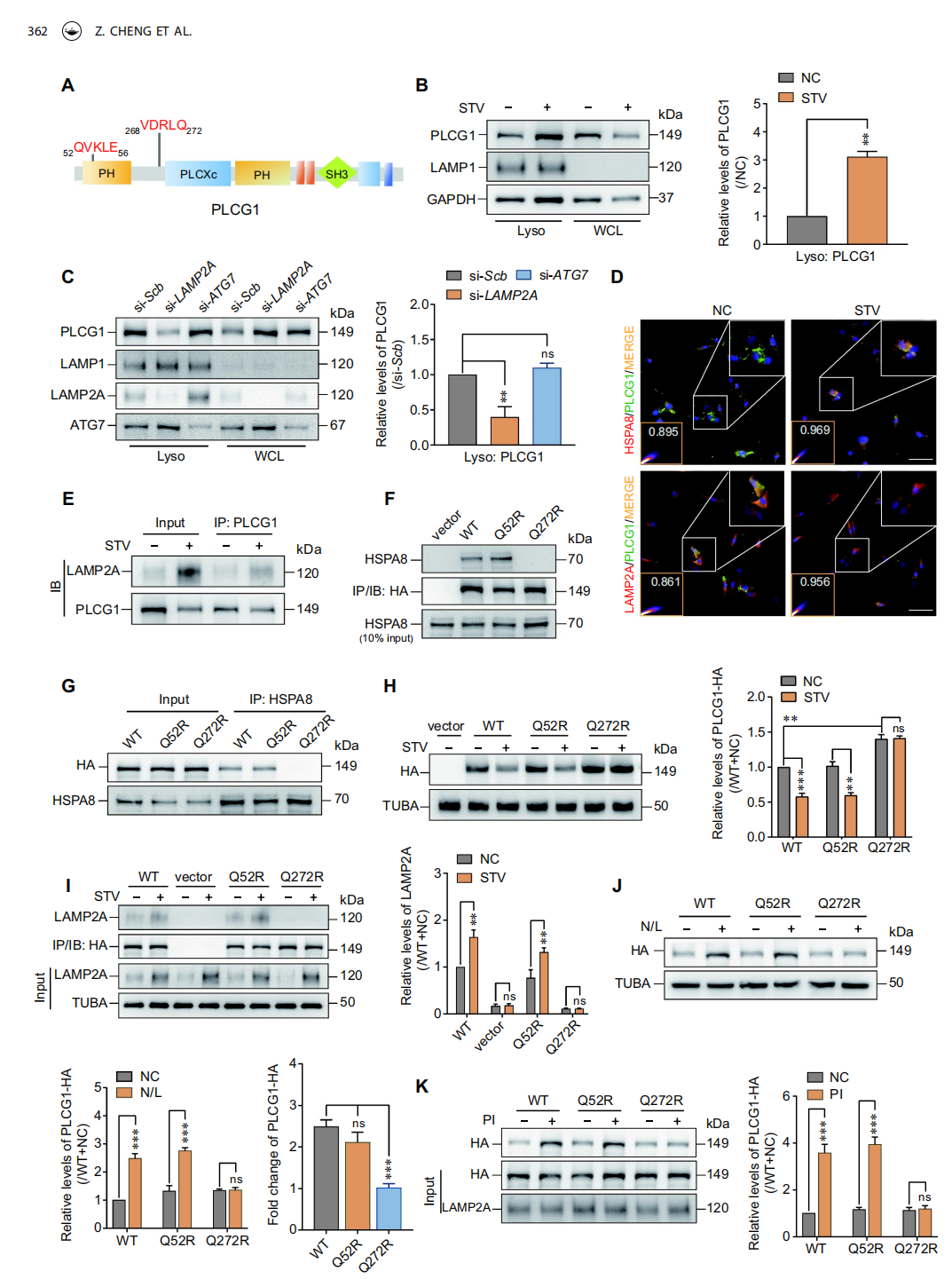
为验证PLCG1是CMA的确证底物,研究发现其含两个保守KFERQ样基序,经实验证实:
268VDRLQ272基序是HSPA8识别PLCG1的关键,该基序突变后,PLCG1丧失与HSPA8/LAMP2A的结合能力,无法发生溶酶体易位,也不能被CMA降解;而野生型及另一个基序突变体均能正常被CMA识别降解。
综上,PLCG1是CMA的确证底物,其降解高度依赖268VDRLQ272基序。
6.磷脂酶Cγ1(PLCG1)以钙依赖的方式诱导细胞衰老
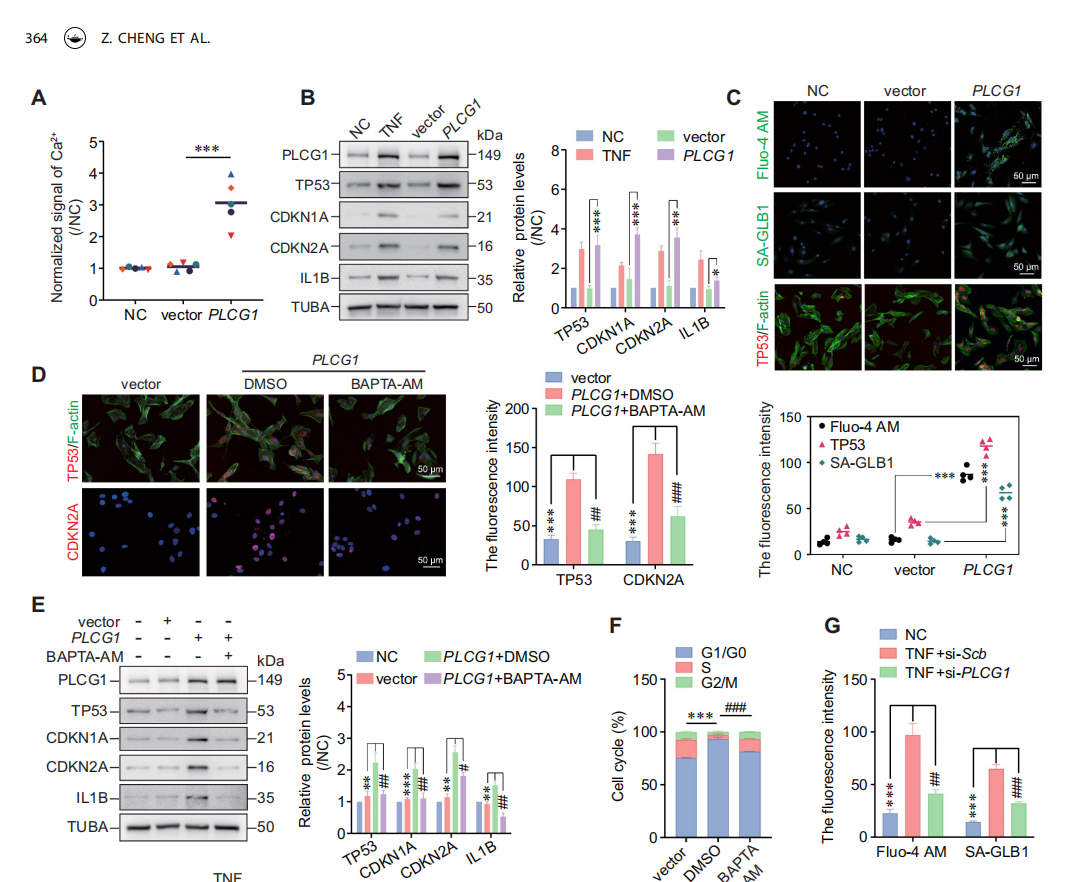
研究探究PLCG1(PLC家族成员,通过IP3信号升高细胞内Ca²⁺)与髓核细胞Ca²⁺水平及衰老的关系,核心实验及结论如下:
慢病毒过表达PLCG1后:髓核细胞Ca²⁺水平显著升高,衰老标志物(TP53、CDKN1A等)、SASP因子IL1B(蛋白及mRNA)上调,SA-GLB1激活,同时细胞S期比例和BrdU水平下降(增殖受抑),ROS水平也升高。
用钙螯合剂BAPTA-AM处理后:上述PLCG1过表达引发的衰老相关现象(标志物上调、SASP激活、细胞周期阻滞、ROS升高)均被显著抑制。
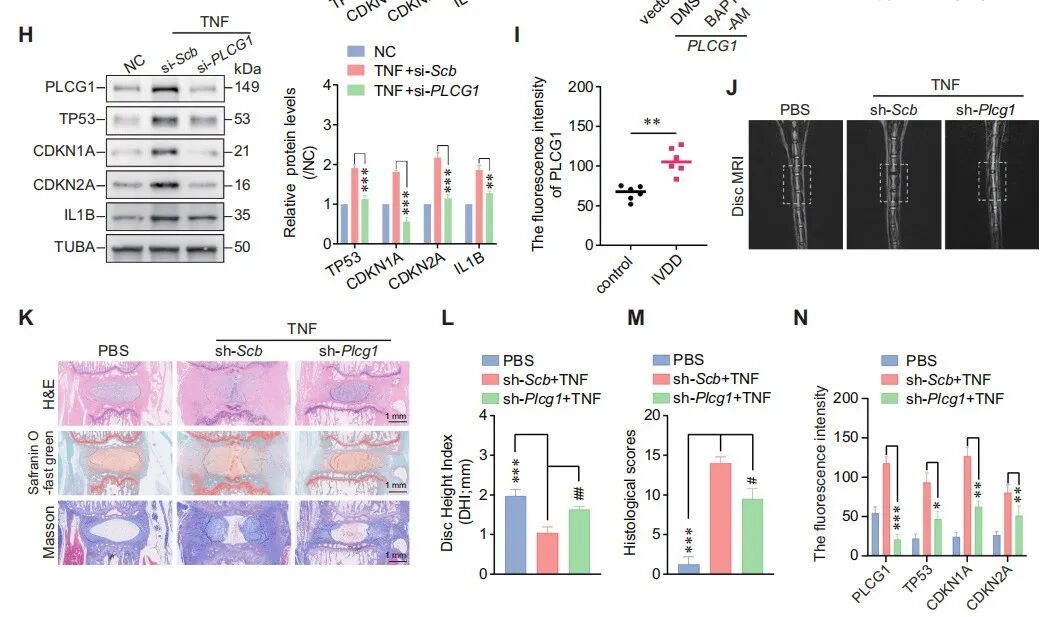
研究证实PLCG1介导了TNF诱导的椎间盘退变(IVDD)中髓核细胞(NPC)的衰老,核心实验及结果如下:
细胞实验:TNF刺激会升高NPC的PLCG1水平,敲低PLCG1可逆转TNF诱导的钙超载、SA-GLB1激活、BrdU水平降低,同时抑制TNF引发的衰老标志物(TP53、CDKN1A等)及IL1B的上调。
临床样本验证:人退变椎间盘组织中的PLCG1水平显著高于正常椎间盘组织。
动物实验:大鼠椎间盘内注射TNF构建IVDD模型,局部敲低Plcg1可明显改善TNF诱导的椎间盘退变,表现为椎间盘高度增加、Pfirrmann评分和组织学评分降低,髓核区域结构更完整,且退变椎间盘中的衰老标志物水平显著下降。
综上,PLCG1是TNF诱导IVDD过程中NPC衰老的关键介导因子。
7.磷脂酶 Cγ1(PLCG1)的异常蓄积介导分子伴侣介导的自噬(CMA)抑制诱导的细胞衰老
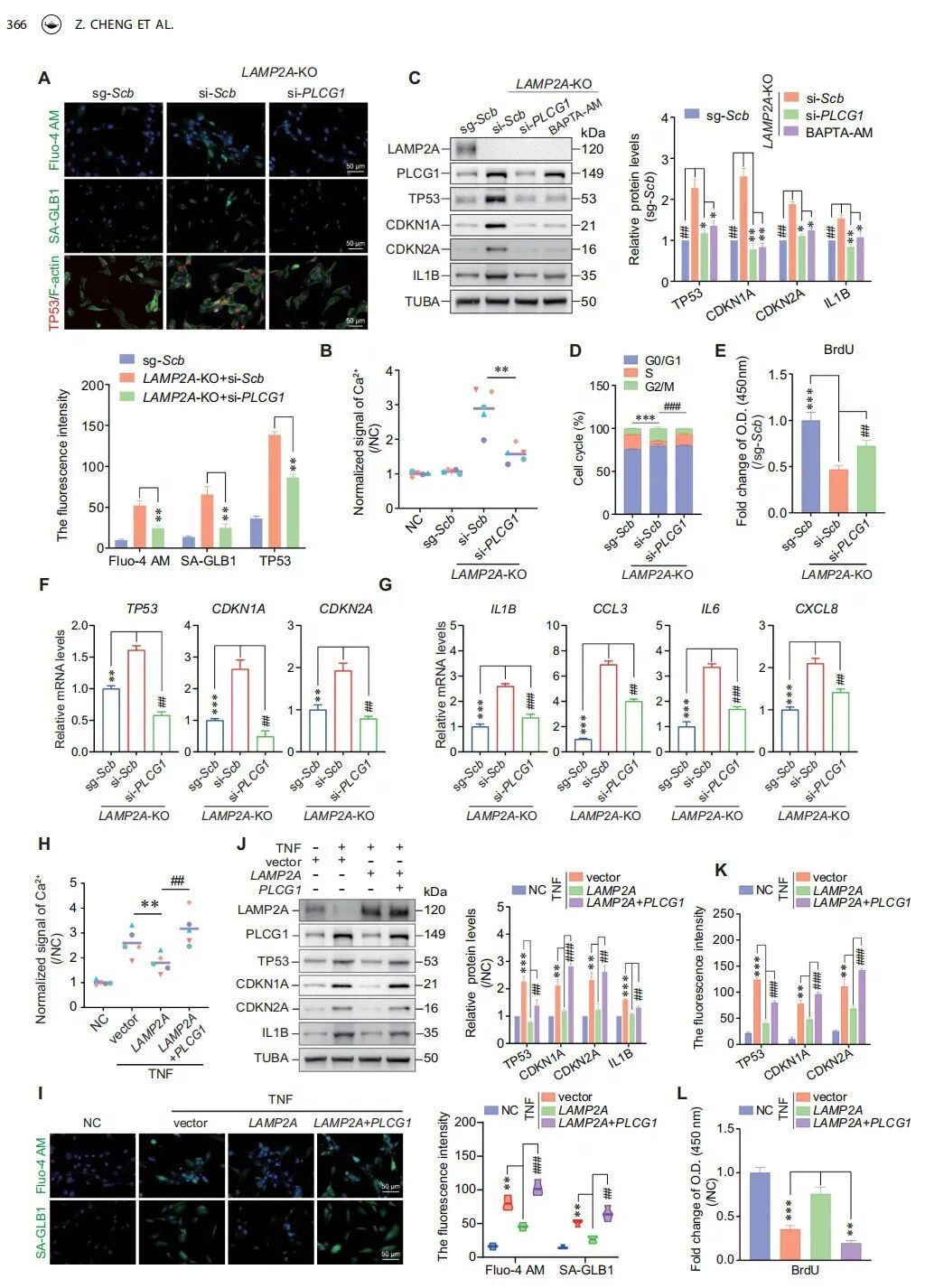
研究进一步验证PLCG1异常蓄积介导CMA抑制及TNF诱导的NPC衰老,核心实验及结论如下:
CMA抑制(LAMP2A-KO)模型中:敲低PLCG1或用其抑制剂,可显著抑制LAMP2A-KO引发的Ca²⁺超载、SA-GLB1激活,逆转衰老标志物及SASP因子的上调,同时恢复细胞周期阻滞,证实CMA抑制时,PLCG1降解受阻是NPC衰老的触发因素。
TNF诱导衰老模型中:单独过表达LAMP2A(激活CMA)可减轻TNF诱导的NPC钙超载、衰老及细胞周期阻滞;但共表达PLCG1后,LAMP2A的保护作用完全消失。
综上,CMA抑制导致PLCG1降解受损、异常蓄积,进而介导了TNF诱导的髓核细胞衰老。
本文围绕TNF诱导的髓核细胞(NPC)衰老及椎间盘退变(IVDD)机制展开讨论,核心是凝练全文实验发现、关联已有研究、明确创新价值,并指出研究局限性与未来方向,具体如下:
1. 核心机制总结:
明确本文关键发现——TNF通过抑制分子伴侣介导的自噬(CMA),导致其确证底物PLCG1降解受阻、异常蓄积;蓄积的PLCG1以钙依赖方式引发细胞内Ca²⁺超载,进而诱导NPC衰老,最终介导IVDD的发生发展**。其中,PLCG1的268VDRLQ272基序是CMA识别并降解它的关键,该基序突变会完全阻断CMA对PLCG1的调控,证实二者相互作用的特异性。
2. 与已有研究的关联与创新点:
结合现有研究,指出本文填补了相关领域空白——此前已知CMA与细胞衰老相关、PLCG1调控钙信号、Ca²⁺超载可诱导NPC衰老,但尚未有研究将CMA、PLCG1与TNF诱导的IVDD关联起来。
本文首次证实CMA在NPC钙稳态调控中的作用,明确PLCG1是CMA的特异性底物,并串联起“TNF-CMA抑制-PLCG1异常蓄积-Ca²⁺超载-NPC衰老-IVDD”的完整通路,完善了IVDD的病理机制网络。
同时,临床样本验证显示退变椎间盘组织中PLCG1水平显著升高,动物实验证实敲低PLCG1可改善TNF诱导的IVDD,进一步佐证了该通路的生理与病理意义。
3. 研究意义:
提出该研究的理论与临床价值——理论上,为CMA在骨骼肌肉系统疾病(尤其是IVDD)中的作用提供了新的研究视角,丰富了PLCG1的调控机制及钙信号与细胞衰老的关联研究;临床层面,PLCG1可作为IVDD诊断的潜在生物标志物,而激活CMA(如过表达LAMP2A)或抑制PLCG1活性,有望成为治疗IVDD的新靶点,为IVDD的精准干预提供了新思路。
4. 研究局限性:
客观指出本研究的不足,例如体外实验主要基于NPC细胞系,难以完全模拟体内复杂的椎间盘微环境;动物模型为急性TNF诱导,与人体慢性IVDD的病理进程存在差异;未深入探究PLCG1下游更具体的钙信号分子机制,也未进一步验证该通路与其他IVDD相关通路(如氧化应激、炎症反应)的交叉作用。
5. 未来研究方向:
基于局限性提出后续探索重点,包括进一步验证CMA-PLCG1-Ca²⁺通路在慢性IVDD模型中的作用;深入解析PLCG1调控Ca²⁺超载的下游分子机制;探索激活CMA或抑制PLCG1的靶向药物可行性,推动该机制的临床转化;同时探究该通路与其他IVDD相关病理因素的协同作用,为IVDD的多靶点治疗提供理论支撑。