【Ucallm】2× Fast SYBR qPCR Mix 产品说明(货号:H7110)
2× Fast SYBR qPCR Mix

产品概述
本产品是采用SYBR Green I嵌合荧光法进行实时荧光定量PCR的专用2×浓度预混液。本产品含有优化浓度的Ucallm Fast HSTaq DNA Polymerase、SYBR Green I、dNTPs、Mg2+、反应缓冲液和稳定剂等成分。主要用于基因组DNA靶序列和RNA反转录后cDNA靶序列的检测。Ucallm Fast HSTaq DNA Polymerase高温加热前,抗Taq单克隆抗体与Taq结合,抑制Taq的聚合酶活性,从而抑制在低温条件下出现的由引物和模板DNA非特异性杂交或引物二聚体引起的非特异性扩增。抗体在PCR反应的预变性步骤中已完全失活,不会阻碍之后Taq酶聚合反应,大大提高了PCR反应的灵敏度及特异性。
优化浓度的SYBR Green I荧光染料掺入DNA双链后,荧光信号增强,而不掺入链中SYBR Green I染料分子荧光信号不变,从而保证荧光信号的增加与PCR产物的增加完全同步,荧光可以在退火或延伸阶段测定。
本产品为2×浓度预混实时荧光定量快速PCR反应体系,使用时只需加入模板、引物、ROX Reference Dye(用以校正孔与孔之间产生的荧光信号误差,根据不同荧光定量PCR仪选择使用)和水,使其工作浓度为1×,即可进行反应。具有快速简便、灵敏度高、特异性强、稳定性好等优点,可最大限度地减少人为误差、节约PCR实验操作时间、降低污染机率。
产品组分
组分货号 |
组分名称 |
规格 |
H7110 |
2× Fast SYBR qPCR Mix |
1ml×10 |
注:不同仪器所需ROX Reference Dye不同,如需添加,需致电本公司或向服务您的销售人员索取:
需加High ROX Reference Dye (50×)的机型: ABI Prism7000/7300/7700/7900HT和ABI StepOne /ABI StepOne Plus。
需加Low ROX Reference Dye (50×)的机型: ABI Prism7500 /7500 Fast, MJ Research Chromo4, Opticon (II), Corbett Rotor Gene 3000。
无需加ROX Reference Dye的机型: Thermal Cycler Dice Real Time System, LightCycler, Smart Cycler System, Corbett Rotor-gene 6000, Agilent Technologies Mx3000P等荧光定量PCR仪。
运输与保存方法
低温运输。-20℃避光保存,保质期24个月,避免反复冻融。如果经常使用,可置于4℃保存至少3个月。
注意事项
使用前请上下颠倒轻轻混匀,尽量避免起泡,并经短暂离心后使用。
尽可能减少2× Fast SYBR qPCR Mix在光下的曝露时间,长时间的曝光可导致荧光信号减弱。
反应液的配制、分装请一定使用无污染的枪头、Microtube等,尽量避免交叉污染。
本品不能用于杂交探针法。
使用方法
用户需自备的试剂:cDNA或DNA模板、引物。
请按照不同品牌荧光定量PCR仪的使用说明书要求进行实验操作。
操作示例:分别以20 µl和50 µl PCR反应体系为例:
1. PCR反应体系的建立:
组分 |
20 µl体系 |
50 µl体系 |
DNA模板a |
1 μl |
1 μl |
正向引物(10 μM)b |
0.5 μl |
1 μl |
反向引物(10 μM)b |
0.5 μl |
1 μl |
2×Fast SYBR qPCR Mix |
10 µl |
25 µl |
High/Low ROX Reference Dyec |
0.4 µl |
1 µl |
Sterile Water |
补足至20 μl |
补足至50 μl |
2. PCR反应条件的设置:
两步法PCR扩增程序
流程 |
温度 |
时间(参考范围) |
循环数 |
预变性 |
95℃ |
2 min(30 s-5 min)d |
- |
变性 |
95℃ |
10s(3 s-15s)e |
40 |
退火/延伸 |
60℃f |
30 s(10s-40 s)g |
|
溶解曲线(仪器自动设置) | |||
三步法PCR扩增程序
流程 |
温度 |
时间 |
循环数 |
预变性 |
95℃ |
2 min |
- |
变性 |
95℃ |
15s |
40 |
退火 |
60℃ |
15-30 s |
|
延伸 |
72℃ |
30 s |
|
溶解曲线(仪器自动设置) | |||
注:以上举例为常规qPCR反应系统,仅供参考。实际反应条件因模板、引物等的结构不同而各异,需根据模板、引物、目的片段的特点设定最佳反应条件,并根据比例放大或缩小反应体系。
3. 在相应的real time PCR仪器上完成实验,并分析结果。
【备注】
本产品仅供科研使用。在确认产品质量出现问题时,本公司承诺为客户免费更换等量的质量合格产品。在所有情况下,本公司对此产品所承担的责任,仅限于此产品的价值本身。
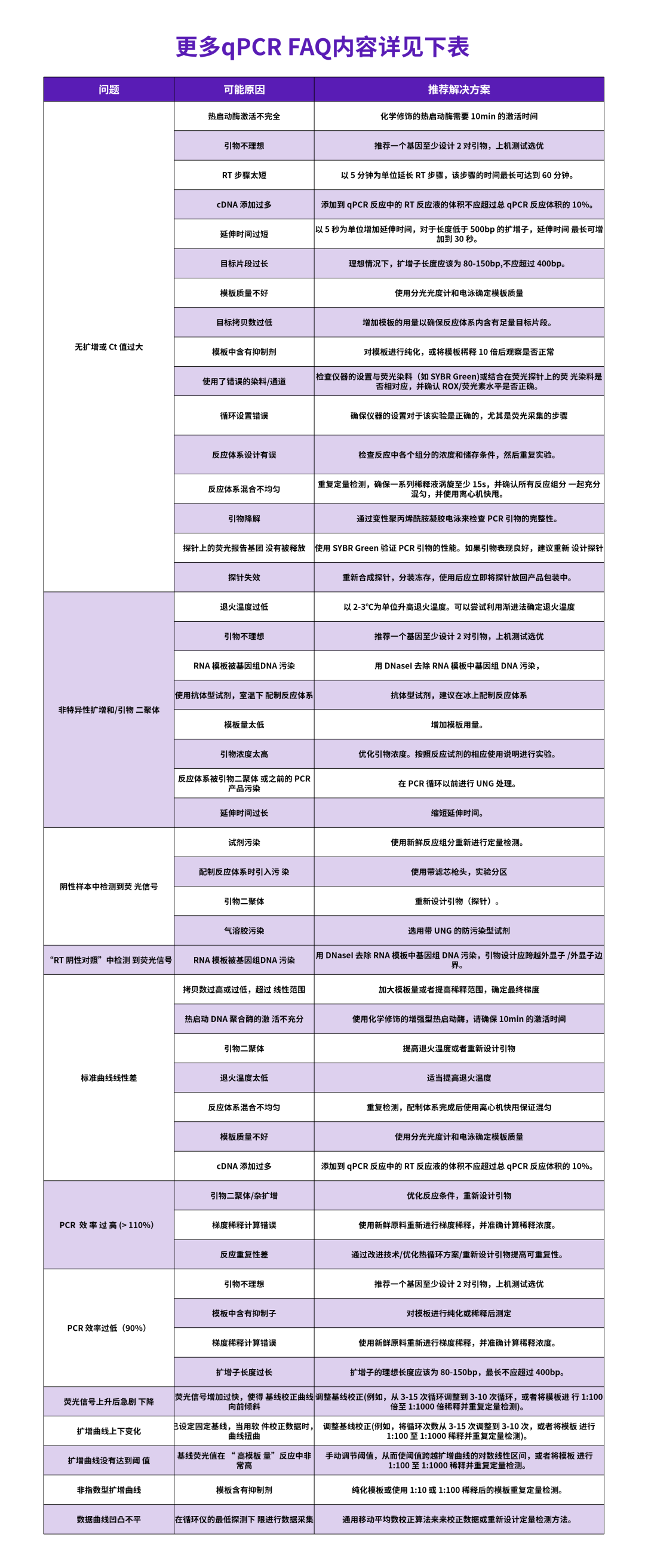
qPCR Mix FAQ
Q
绝对定量与相对定量有哪些差别?
A
绝对定量与相对定量有如下差别:
1. 绝对定量:是对未知样品的绝对量(拷贝数)进行测定的方法;通常应用于病毒、细菌、衣原体、支原体的定量检测及转基因食品的检测。
2. 相对定量:是分别测定目的基因和参比基因的量,再求出对于参比基因的目的基因的相对量,最后再进行样品间相对量的比较。主要用于检测细胞 mRNA 表达量的变化;比较不同组织的 mRNA 表达差异;验证基因芯片、siRNA 干扰的实验结果。
Q
如何选择制作标准曲线的标准品?
A
为了保持与实际检测样品间的扩增效率的一致性,作为标准品应尽量选择与实际检测样品结构近似的样品。例如以基因组 DNA 为起始材料时就要选择基因组 DNA 作为标准品,进行mRNA表达解析时最好选择表达目的基因的Total RNA(或以Total RNA为模板合成的cDNA)作为标准品。即使扩增碱基序列相同,但整体模板不同,也有可能导致PCR扩增效率不同(比如基因组DNA和质粒DNA等)。
Q
进行Real Time PCR实验时,需要做哪些确认工作?
A
进行Real Time PCR首先要确认的工作如下:
1. 引物和探针的序列是否有错误?组合是否正确?
2. Total RNA是否有分解的可能?
3. 各种试剂是否忘记加入?
4. PCR条件和荧光检出步骤是否设定正确?如果有成功反应经历的模板(PositiveControl),设置Positive Control反应,很容易进行以上项目的确认。Total RNA一定要通过电泳和OD分析确认其质量。
Q
如何确认模板中是否含有阻害物质?
A
有时Total RNA或cDNA中含有对反转录反应和PCR反应的阻害物质(如RNA提取时使用的有机溶剂等)。为了确认这样的阻害物质的有无,使用较高浓度的模板按3~4个梯度稀释,并使用其进行RealTime RT-PCR反应或RealTime PCR反应。如果无阻害物质存在,得到的Ct值会依存模板浓度的变化而变化;如果有阻害物质存在,就会发现高浓度的模板有反应性能下降的现象。
Q
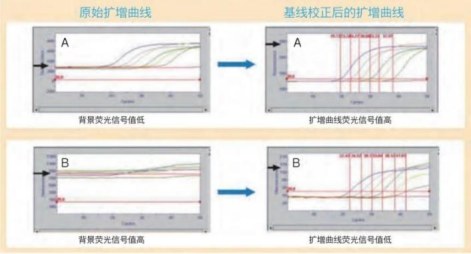
扩增曲线的荧光信号值低的原因是什么?
A
扩增曲线存在问题时,首先应确认基线校正或ROX校正前的原始曲线,扩增曲线的荧光信号值低多数是由于背景荧光信号值过高造成的。如下图所示,A的原始扩增曲线背景荧光信号值约为280,其基线校正后的扩增曲线显示较高的荧光信号值(约500);而B的原始扩增曲线背景荧光信号值可达800,其基线校正后的扩增曲线荧光信号值较低(约150)。使用嵌合荧光法进行检测时,背景荧光信号偏高大多数是由于模板量过高,染料嵌入到初始模板DNA中发出荧光造成的。使用荧光探针法进行检测时,背景荧光信号偏高大多是由于设计的探针质量差使淬灭基团的淬灭作用不充分、报告基团和淬灭基团的搭配不合理、探针的荧光标记效率低等原因造成的。
Q
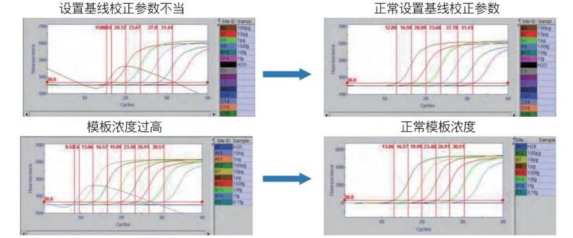
出现扩增曲线朝右下方下落现象的原因是什么?
A
出现这种现象一般有两种可能,如下图所示。
1. 由于基线校正不恰当。确认设置的基线校正范围的参数,按仪器说明书要求重新设定再进行解析。
2. 使用的模板量过多,扩增曲线过早起峰,使基线校正不能正常进行。当扩增曲线在
10 Cycles以内起峰时,要将模板稀释100~1000倍后使用。
Q
PCR扩增效率低的原因有哪些?
A
由标准曲线的斜率计算得到的PCR扩增效率低时,可以考虑以下几点原因。
1. PCR反应性能差。引物、试剂、PCR扩增条件需再摸索。
2. PCR阻害物质的混入。模板提取方法的再摸索。
3. 标准品稀释不准确。使用专用标准品稀释Buffer。稀释的低浓度标准品常常有易分解不稳定的现象产生,所以使用的稀释液中最好加入与实验材料不同生物种的tRNA和rRNA以起保护作用。但偶尔也有因加入的tRNA和rRNA的序列与目的基因序列具有同源性而产生的干扰现象。
Q
PCR 扩增效率过高的原因是什么?
A
使用嵌合荧光法检测时,有时非特异性扩增会导致PCR扩增效率高,要通过融解曲线分析加以确认。
Q
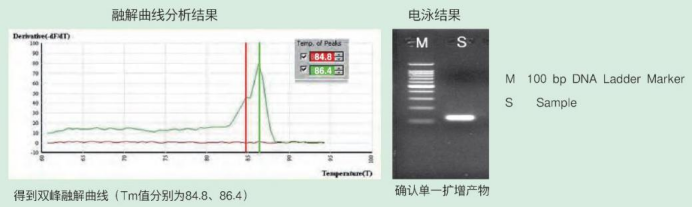
融解曲线分析有多个峰产生的原因有哪些?
A
首先怀疑发生了目的片段以外的扩增。
1. 引物二聚体等非特异性扩增。优化PCR条件或再设计引物。
2. 存在目的基因的Variant。获取Variant信息,再设计引物。
3. Genomic DNA来源的扩增。DNaseI处理或考虑基因组结构、引物再设计。
但也有偶尔例外的情况。那是由于目的序列内部GC含量等的不均一造成同一扩增产物解离稍有偏差,致使虽然为特异性扩增,但融解曲线分析却出现非单一峰型的情况,但此时电泳确认结果却为单一条带(如下图所示)。因此,对初次使用引物的扩增产物有必要进行电泳确认,其目的有两个:①单一峰型时可以确认扩增片段大小,判断是否为目的产物;②非单一峰型时也有必要对扩增产物进行确认,如果电泳结果显示为单一条带,并与目的片段大小吻合,可以判断为进行了特异性扩增。
Q
qPCR实验中都有哪些对照,作用是什么?
A
我们可以按照下面的表格设计对照实验,从而更好的分析相关数据
对照 |
目的 |
Negative control: No template (NTC) |
检验是否有模板污染 |
No reverse transcriptase (NRT) |
检验RNA 样品中是否有DNA 污染 |
No amplification control(NAC) |
检验是否有聚合酶污染 |
No probe control(NPC) |
检验荧光污染 |
Buffer: Only contains buffer |
检验背景荧光 |
Q
正常的扩增曲线应该是什么样子的?
A
A 、曲线平滑;起峰时间正常;NTC 和 NRC 无扩增(起峰晚);Cq 值一般为 15—30 之间。
Q
样品Cq值大于30,该如何对待?
A
对于 Cq 值 30-35 的样品可以通过 NTC 的情况来判读结果是否有效,一般来说 NTC 无扩增 或者 Cq(NTC)-Cq(simple)>5;而大于 35 的 Cq 值可以认为是无扩增。


点在看,传递你的品味